天然有机化合物结构的精准确定是天然药物化学的基本任务之一,提供正确的分子结构对后续的药物化学、化学生物学、合成化学和生物合成等研究至关重要。随着NMR、MS以及量子化学计算辅助ECD等技术的发展,复杂天然产物结构解析的正确率已经得到了大幅度的提高。然而,面对含有新颖氮杂环的高度复杂的海洋生物碱类化合物的结构解析仍然具有巨大的挑战,特别是在利用具有经验性的NMR数据,在常规思维定势下进行结构解析时,往往会推测出看上去合理、事实上错误的结构。
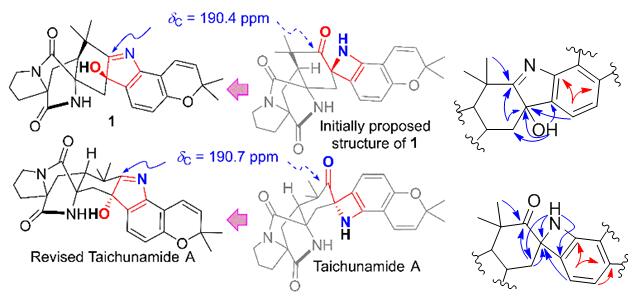
海洋药物教育部重点实验室顾谦群课题组李德海教授等人从红树林来源的真菌Aspergillus versicolorHDN11-84中分离得到一个结构新颖的复杂多环生物碱Taichunamide H,其核磁数据同2016年报道的Taichunamide A(Angew. Chem. Int. Ed.2016, 55, 1128 –1132)具有高度的相似性,因而最初研究人员将该化合物解析为Taichunamide A的非对映异构体(结构1a)。在进一步确定Taichunamide H的绝对构型时,研究人员发现其TDDFT-ECD曲线和13C NMR化学位移计算值同实验值不能很好的吻合;化学位移计算中最关键差异是190.4 ppm处的季碳。一般在结构解析中,化学位移值190 ppm以上的季碳首先会被认为是羰基碳;而基于这一归属,Taichunamide H会根据其它NMR数据“合理的”解析出其中的氮杂环丁烷(spiro-azetidine)片段。同时,考虑到这种自然界中非常罕见的环系可能不会稳定存在,而实验中该化合物却相对稳定,这一理化性质也并不吻合。因此对结构进行了重新解析,并最终通过X-单晶衍射确定了正确结构(结构1);阐明结构中190.4 ppm的季碳应该归属为碳氮双键的亚胺碳。此外也修订了Taichunamide A的结构(revised Tai-A),并经过量子化学计算得以佐证。
以stephacidins、notoamides和versicolamides等为代表的bicycle[2.2.2]diazaocatane复杂多环生物碱近年来一直是生合成以及全合成的热点;其中在stephacidins/notoamides等结构中,以亚胺形式存在的吲哚片段N原子上通常会连有O或者OH等吸电子基团,从而导致亚胺碳的化学位移在140-150 ppm左右;而在Taichunamides A和H中,碳氮双键N上却没有这种吸电子基团,导致亚胺碳化学位移出现在了190 ppm以上,这一异常化学位移具有较高的误导性,应引起高度重视。
该研究成果已经在线发表在JCR小类学科一区Top期刊Org. Lett. 2018, DOI: 10.1021/acs.orglett.8b00061。海洋药物教育部重点实验室2014级制药工程硕士李凤为本文第一作者。